
2023-10-08 来源 : 英勇向前

2023年10月5日,FDA关于Sotorasib的ODAC会议结束,专家组以2票赞成,10票反对的结果,认为CodeBreak200的主要研究终点无法被可靠地解释。
FDA指出当确证性研究无法证明获益时,并不会自动撤销FDA加速批准(AA),最终决定将基于研究总体获益、药物安全性优势、以及当前而非AA批准时的风险获益。
FDA认为CodeBreak200研究PFS获益小且无OS差异,更重要的是,出现诸多潜在的系统性偏倚以及研究执行问题,因此召开本次ODAC会议。
系统性偏倚信号的出现,及其对研究整体的影响如下。
回到CodeBreak200研究,PFS获益仅5周(mPFS 5.6 vs. 4.5)短于影像间期的6周,即患者在发现PD与前一次非PD之间的任何时期均可以已经发生了PD。FDA使用interval censoring分析,Sotorasib相比多西他赛的PFS获益仅5天。
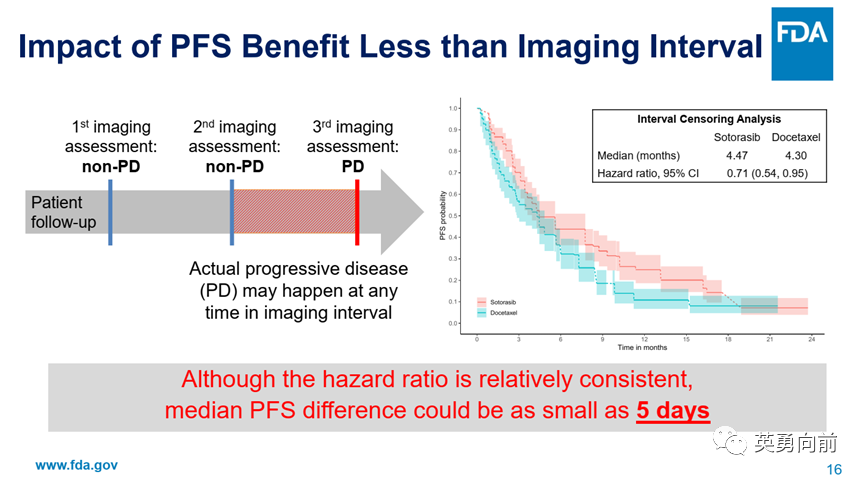
FDA认为CodeBreak200研究存在的问题包括:
非对称性的早期退组。随机后未治疗患者在两组分别为2(1%)和23(13%),即,多西他赛组23例患者未接受治疗。这打破了随机的均衡性,假设退组的患者有更好的预后,则目前的PFS数据将会被高估。
Amgen:分析23例患者存在更差预后的多种因素,例如ECOG评分、肝转移等。采用统计学方法,协变量调整分析,以及20,000次simulation后仍证明治疗组优于对照组。
研究者评估利于治疗组。研究中的早期/晚期不一致不均衡,说明从研究者主观角度看,他们‘希望’多西他赛组患者早点退出,而sotorasib患者多接受一段时间治疗。

基于研究者评估的早期交叉换组影响BICR的评估。19例换组由于早期换组导致数据删失,假设这些换组的患者(数据被censor)相比BICR后换组的换组状态更好,则目前的PFS还是被高估的。

FDA认为申办方引入了确认进展评估(confirmation of progression,COP),COP由BICR供应商提供,由独立于研究的人员进行。
COP的本意在于给研究者的判断提供即时的第二视野,如果使用恰当不会对BICR发生任何影响。但申办方可能使用COP的结果稽查BICR判断,使BICR调整了12例PFS的事件,这导致HR从0.8改善到0.70。
这与BICR规则相悖,因此有错误使用COP的嫌疑。

FDA列举出CodeBreak200研究中可能存在的问题。对于开发标签此类研究,无法要求完美的执行,但为了提高研究质量,
申办方应:1. 研究者/患者教育,2. 允许交叉患者;3. 实时BICR;4. 终点选择(PFS vs. OS);5.关注OS数据的收集,即便患者退出治疗。

参考:October 5, 2023: Meeting of the Oncologic Drugs Advisory Committee Meeting Announcement - 10/05/2023 | FDA
Sotorasib和Adagrasib的获批使Kras成为近几年的热门,当然这只是开始。后续探索围绕:靶向KrasG12C的advance-in-class类药物、联合治疗、疾病阶段、对G12D/G12V/G13V等困难靶点的开发。
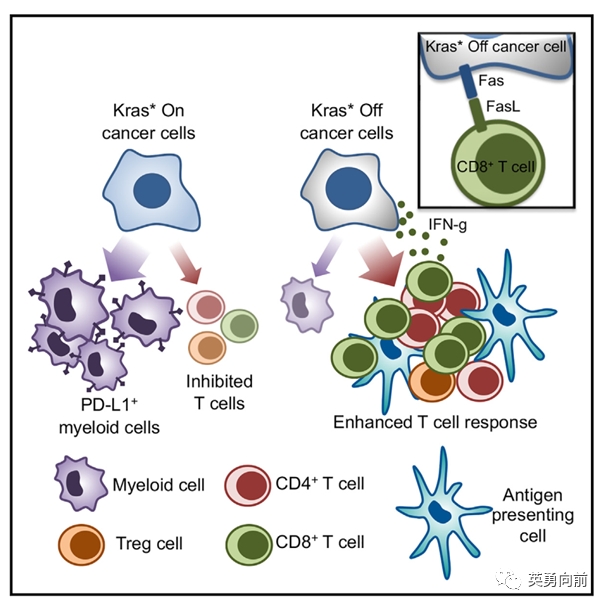
9月11日,Cell杂志发表,在胰腺癌动物模型中观察到Kras G12D与免疫抑制状态的相关性。简单来说,清除Kras G12D可上调肿瘤细胞Fas受体、增强肿瘤微环境中CD4+和CD8+的T细胞浸润。
理论上,Kras G12D联合免疫治疗协同增效,结合现有证据联合上游EGFRi也会发挥联合增效作用,等着Kras G12Di成药吧。
8月24日,罗氏的Kras G12C抑制剂Divarasib在NEJM公布临床I期数据。137例患者,60例NSCLC、55例CRC、22例其他肿瘤。NSCLC的cORR为53.4%(39.9,66.7%),中位PFS为13.1个月;CRC的cORR为29.1%,中位PFS为5.6个月。

9月世界肺癌大会上,Sotorasib和Adagrasib更新临床数据。
Sotorasib的CodeBreak 101研究评估联合化疗治疗NSCLC,入组20例1L患者,cORR为65%,DCR为100%;13例2L患者,ORR为54%,结果与PD-L1的表达无关。
Adagrasib公布Krystal-1研究,132例2L+
NSCLC患者。ORR为43%,PFS为6.9个月,OS为14.1个月,2年OS为31.3%。相比获批时112例患者数据,ORR为43%,DoR为8.5个月,PFS为6.5个月,OS为12.6个月,疗效持续且增强。
值得强调的是,adagrasib对脑转移患者具有独特疗效,基线存在脑转移者(n=26)OS为14.7个月,与总体人群无差别。其次肝毒性较轻,成为探索联合pembro用于1L NSCLC的重要前提。
Sotorasib和Adagrasib均是通过单臂研究获得FDA加速批准(AA)的。对于AA的要求,3月FDA发布了指导规范草案,进一步提高对AA的要求。特别强调确证性三期研究的对AA审批的支持价值,但仍保留单臂支持AA的通路。
2023年8月,CDE发布关于附条件批准的征求意见稿。
总结几点:
1. 某药品附条件批准后,原则上不再同意同机制、同靶点、同适应症的同类药物申请附条件;
2. 对确证性研究提出具体要求,沟通时需要启动三期,原则上不超过4年完成,并在附条件批准上市后每12个月书面报告研究进展;
3. 允许沟通后合理调整研究方案,如无法按期完成确证性研究的,存在暂停销售风险。
非常明确的是,全球监管机构对于抗肿瘤药物的加速批准在不断收紧。
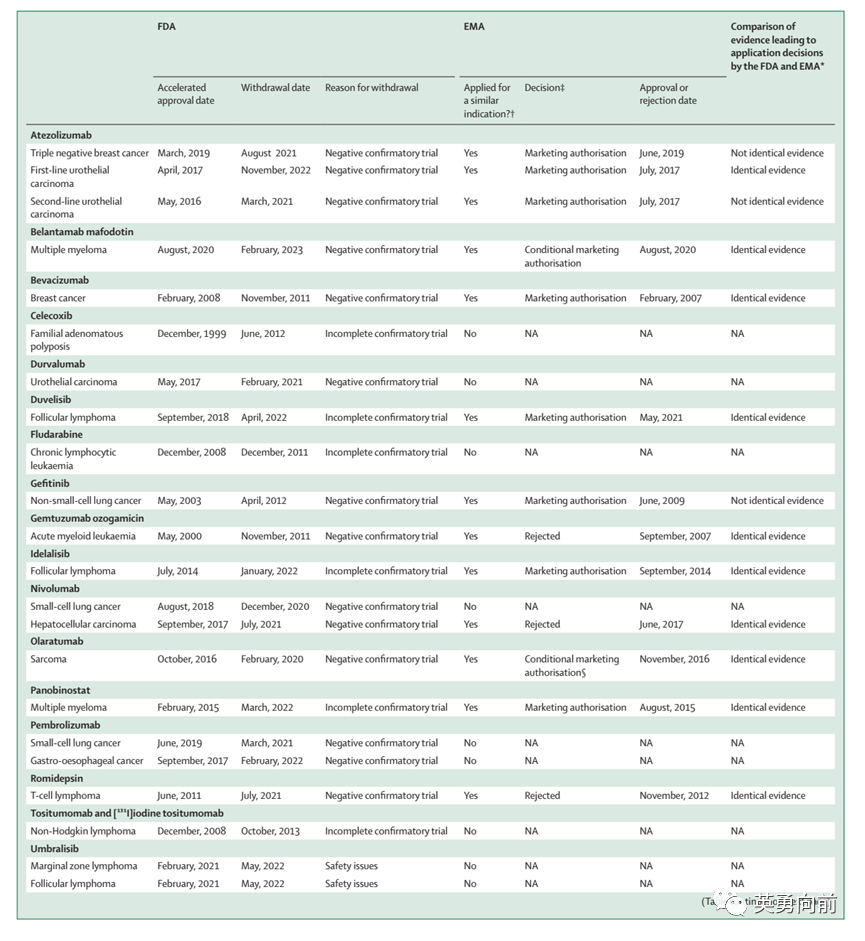
2023年9月,Lancet杂志发表文章对比了EMA和FDA在AA撤回上的异同点。

文章分析1992年至2023年4月,FDA批准的肿瘤药物中114个加速批准(AA),其中23个(20%)适应症(18个药物)被撤回,17个(74%)发生在最近3年里,撤回的主要原因包括确证性研究失败、无法继续完成研究,或安全性风险。
同期EMA来看,23个适应症中,13个(57%)在EMA有相同的适应症申请,其中3个被拒,1个获批后被撤回,其他9个在EMA仍在获批状态。
例如,Duvelisib基于单臂研究于2018年9月获FDA的AA批准,但由于三期失败于2022年4月从FDA撤市。然而,EMA基于同样单臂研究于2021年5月常规批准,无后续动作。Idelalisib有相似的情况。
EMA撤回率远低于FDA,究其原因,作者认为,
1. 审批路径不同,如EMA给予了常规批准后难以撤回;
2.监管对benefit-risk的考虑不同;
3. 新药递交的时间不同,FDA递交往往先于EMA,这可能导致结果不同。
在各个国家监管部门在不断提高要求的同时,9月14日NEJM杂志发表了对这一问题的专家观点,但我个人认为这些观点过于苛刻,甚至偏颇。

1.专家建议FDA关闭单臂研究支持AA的通道。该通道应该仅用于特殊案例,例如罕见肿瘤缺少有效治疗,即便如此,也应该以CR率作为判断标准,而非ORR,因为PR无法可靠地体现药物活性。
个人观点:这一表述几乎否认了过去30年里基于单臂研究AA的价值,尽管少部分适应症被撤回,但不应该否认AA原本的价值,即,为晚期癌症患者提供提前接受基于替代终点的新药治疗。
这些AA在后续的确证性研究中,大多获得临床获益的确证。与此同时,加速审批路径也孵育了突破性新药创新的生态系统,为社会带来繁荣和就业机会。
针对CR作为终点,不应一概而论。CR指在现有可靠的检测手段下,肿瘤消失。但肿瘤消失并不代表治愈,例如白血病的原始细胞<5%就符合CR标准之一,淋巴瘤的PET-CT转阴也是CR,实体肿瘤则需要影像学的肿瘤消失,但CML却已经来到了深层MRD水平。
值得反思的是MDS。在CD47案例中,前期HR-MDS的数据提示相比AZA单药,联合CD47单抗,CR率出现2-3倍的提高,后来国内外涌入了大量的‘创新’药。2023年7月21日和9月26日,Gilead分别暂停了HR-MDS和AML-TP53的两项国际多中心三期研究。
主要原因是前期高估了二期数据,特别是CR在MDS中的临床价值。
MDS是一组癌前癌症,原始细胞本就不高,其疾病主要矛盾是单克隆性、病态造血和转化为AML风险。对其评估的方法又是2006年的IWG,CR的参考价值是非常有限,换句话说,没有CR也不影响生存。
例如AZA中国的桥接研究中,仅1/72例患者出现PR/CR,但并不影响AZA本身的临床价值,这和疾病的特殊性有关。
时至今日,CD47又要从头来进行临床探索,风大雨大,真的很难!
2. 专家认为PFS无法真实反应临床获益,和生存质量。当一个药物被AA批准后,应该要求OS和/或生存质量评分作为powered的终点(对应样本量)。
个人观点:PFS捕获进展和生存数据,不可能没有意义。在淋巴瘤、骨髓瘤等大部分的血液肿瘤里,PFS几乎是唯一的主要终点,类推在其他生存时间较长的肿瘤中,PFS也是较为理想的指标。
然而当药物被AA批准后,在盲态下以OS为终点,或至少是powered的次要终点或是合理的。
3. FDA强调研究应该展现治疗的临床意义,而非单纯统计学意义。但如何定义临床意义尚缺乏客观指标。
专家认为欧洲使用的“the Magnitude of Clinical Benefit Scale”值得推荐,这一量表不仅考虑生存时间,也同时考虑了药物本身的毒性-效应谱,以及患者生存质量的改善等指标。相对较为客观。
4. 专家认为FDA对于三期研究里对照组要求 ‘合理的可用治疗’,此处 ‘合理的’比较不合理,因其太具主观性。
专家建议选用 ‘最佳治疗 – best available
treatment’。对于informative
censoring(由于AE或缺乏疗效、亦或者交叉换组、接受方案外治疗等情况,导致两组的drop-out不均衡的),FDA也应该提供相应的指导意见,以保证研究方法的严谨性。
个人观点:针对最佳治疗的定义存在差异,个人认为对照组的选择应该体现广泛使用的原则。如果只是以三期研究的阳性结果作为“最佳治疗”的判断标准,显然是非常不合适的。
临床研究结果无法代表广泛人群的使用结果,并且难以迅速广泛推广。任何新的治疗一定有利有弊,因此应该根据具体情况加以判断。
以治疗DLBCL的R-CHOP方案为例,尽管Polivy已经获批,但现在三期研究中对照组的选择,应该用R-CHOP而非Polivy+R-CVP,因为后者并没有太多的临床证据,全球也未广泛使用。
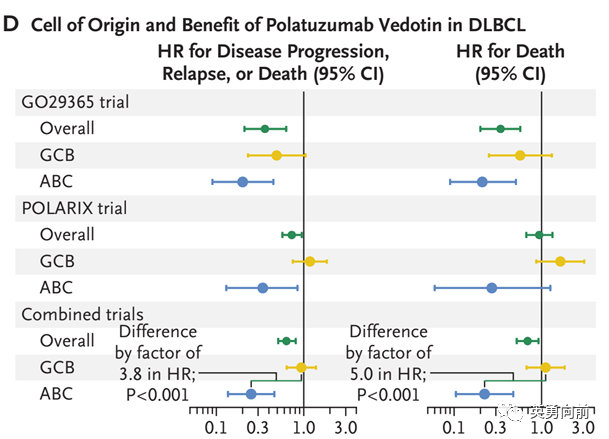
进一步分析,Polivy的治疗结局与细胞起源(COO)有关。

在POLARIX研究中,ABC亚型患者的PFS的HR为0.34,OS为0.27,远高于GCB亚型的1.18(PFS)和1.64(OS)。
POLARIX研究收集到668例(共879例)患者COO数据,ABC亚型在Pola-R-CHP组为102例,对照组R-CHOP为119例,COO使用NanoString方法检测。
尽管结果看起来很坚实,但我仍保持怀疑。在一个差不多221例患者的研究里,见到了HR为0.34和0.27,真的有那么好么,原因是什么?
来那度胺的ROBUST研究入组570例ABC患者,使用完全一样检测方法,带着坚固的早期临床数据,一样失败了,而失败的主要原因,我认为是对照组大幅优于预估,而非治疗组不好。
这是临床研究的魅力。
5. 专家认为FDA没有指出当三期研究无法确证获益时,该如何处理。自2020年之后,FDA开始通过召开advisory committee的形式讨论,大约20个适应症因此被撤市。专家因此建议FDA应考虑设立自动撤市机制,自动匹配失败的确证性研究。
个人观点:自动撤市是不合理的。临床研究是自然和社会科学的集合体,应该针对具体情况加以判断。
只要决策的流程公平、科学、透明,就是合适的。不应该自动撤市。
2023年9月,FDA发布如何使用单个研究来展现关键证据。我认为这是关于如何理解药物开发的理论性的逻辑构建,非常值得学习。

FDA指出一般情况下需要两个充足、良好控制的研究,相互佐证提供疗效确切的证据。而对于单个研究,存在发生偶然性的可能。
因此,应考虑:
1. 临床证据与适应症的相关性,例如药物已获批治疗某种感染,而不同部位的感染则差异不会太大。
2. 药物机制的阐述,必须明确药物针对哪种机制发挥作用,避免了从‘果’反推‘因’的可能。果,应该单独来自于某个‘因’,起码是强关联。
3. 动物研究结果一致性。
4. 同类机制药物的开发情况。
5.疾病的自然史。
6. 临床证据。
上述完整的逻辑线是开发新药的基本要素。如果仅凭单研究的阳性结果,而缺乏上述完整的逻辑线,无法说清因果关系,这对监管的批准会产生问题。
另外,FDA在9月底发布对实验室开发诊断(LDT)的监管,即,明确即便体外伴随诊断(IVD)是实验室生产的,但仍需要被纳入IVD进行监管;FDA打算逐步取消对LDT的自由裁量,使LDT纳入到IVD相同的监管体系中。
简单来说,LDTs在美国将失去存在的合法性,必须将原有的应用逻辑推到重来,以适应新监管,FDA将覆盖医疗保险和辅助服务中心(CMS)的LDT监管权。

回到肿瘤的新药开发,肿瘤的免疫治疗仍然是热门。PD-1取得巨大成功后,肿瘤免疫进入T细胞衔接剂(TCE)和CAR-T阶段。
TCE和CAR-T首先在血液肿瘤中取得重大进展,主要原因是独特的靶点选择,提高患者on-target off-tumor的耐受性。
换句话说,B系肿瘤的独特靶点包括CD20、CD19、BCMA等,而B系清除后的on-target off-tumor效应也就是抗体缺失,除了COVID这样难缠的病毒,大部分患者的耐受性较好。
B系肿瘤包括B-白血病、B-淋巴瘤和骨髓瘤。B-NHL里,目前已批准三个CD20*CD3的TCE,而第四个TCE(Odronextamab)的PDUFA在安排在2024年3月底(优先审评)。除了淋巴瘤,骨髓瘤也是遍地开花。

对比骨髓瘤治疗中的TCE和CAR-T。TCE的优势在于现货供应、CRS发生率低。而CAR-T的优势在于一次使用但CRS相比较高,有10%的生产失败率,生产周期大约4-6周。
特别值得一提的是本文列举的teclistamab(BCMA-CD3),每周一次给药,ORR为63%,中位PFS为11.3个月,CRS较低,但严重感染发生率(3级以上)高达45%,明显高于一般CAR-T的20-30%。
感染在临床上要格外重视,当患者使用TCE时,应升级预防感染的标准,特别强调对(机会性)感染的预防和及时有效的治疗,起码要和白血病化疗后的重视程度相仿。

除CRS和感染外,TCE和CAR-T治疗还要注意血小板的降低(或全血降低),这可能与细胞因子攻击骨髓有关,也或许是一种慢性的CRS表现。
2023年7月,我国学者发表论文,提出血小板降低的四种影响因素:基线血小板水平、基线血红蛋白、基线铁蛋白、CRS的发生。
该研究有指导临床使用和研究设计的价值。

南京传奇生物的BCMA-CAR-T临床三期CARTITUDE-4研究结果,于7月27日发表于NEJM,419例RRMM患者随机接受Cilta-cel和医生选择治疗。

中位随访15.9个月,中位PFS分别为未达到和11.8个月(HR
0.26),12个月的PFS率分别为75.9%和48.6%,CR率为73.1%和21.8%,MRD为60.6%和15.6%。大部分患者发生CRS但3级和以上为1.1%,严重感染并不是主要的问题。
传奇的数据,合作的典范,患者的获益。
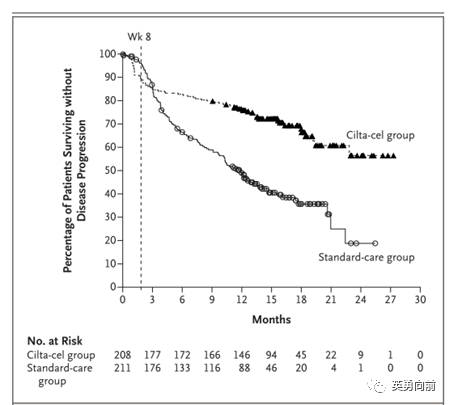
CAR-T治疗大B细胞淋巴瘤的ZUMA-7三期研究,针对的是早期复发或难治的患者,随机359例患者接受axi-cel和标准治疗。
随着随访时间延长到47.2个月,预估4年OS为54.6%和46%(HR
0.73)。
在CAR-T治疗后,患者的B细胞水平在第3-6个月为最低值,2年后逐步恢复。而CAR-T细胞水平逐步递减,至24个月几乎消失。
分析和OS相关因素发现,回输后28天内的CAR-T细胞水平与OS无关,而相关因素包括高比例记忆性T细胞表型(CCR7+CD45RA+)和低比例效应性记忆细胞(CCR7-CD45RA-)。
Axi-cel的感染率为44.7%(3级和以上16.5%),B细胞清除率为62.3%(3个月)和22.6%(24个月)。

CAR-T在白血病、淋巴瘤和骨髓瘤中的成功,在于B系作为治疗靶点的独特性,其独特性在于广泛表达于肿瘤细胞表面、正常细胞被清除后的问题可以解决、肿瘤异质性较小等因素。
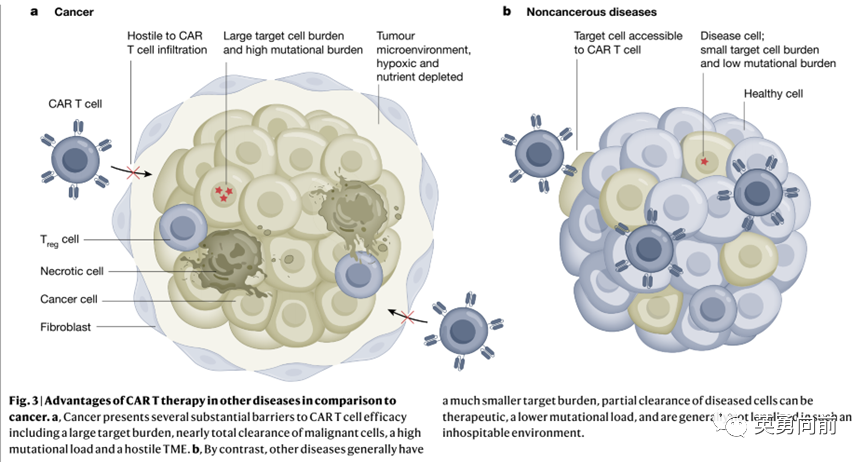
CAR-T在实体肿瘤中探索是令人失望的。首先的原因是缺少类似B系的理想靶点(CD19/20/22/BCMA等)、实体肿瘤异质性大、存在肿瘤微环境抑制CAR-T发挥效应。
肿瘤的清除追求的是‘全或无’,去除一部分的肿瘤将存在复发风险。
基于这些考虑,Carl H. June于7月在Nature杂志发表观点,认为CAR-T是时候进入非肿瘤领域了。

相比实体肿瘤,非肿瘤性疾病的疾病负荷更低,清除部分异常细胞便可能发挥治疗作用。
这些特点很适合探索CAR-T的治疗。这些非肿瘤性疾病包括自身免疫性疾病、纤维化、感染性疾病、衰老性疾病等。
目前已有少许临床数据用于SLE和其他免疫性疾病,取得不错的结果,可以特别关注。
细胞治疗在实体肿瘤中进展较快的有小细胞肺癌(SCLC)和胃癌。
SCLC是一类高度侵袭性的肿瘤,具有高增殖率、治疗反应率高但容易复发和转移等特点。预后不好,5年OS约7%。除化疗以外,PD-L1的获批是仅有的新药成果。
然而,DLL3作为潜在的治疗靶点是SCLC近几年的热门。
9月11日,Amgen公司的DLL3-CAR-T(AMG119)发表早期临床药理数据。
AMG119的起始剂量3*105个CAR-T细胞/Kg,后剂量递增。在给药患者中,观察到1例PR。细胞扩增更容易见于<59岁的年轻人群,而与治疗线数无关。

当然,DLL3作为靶点的探索还包括ADC、TCE。个人以为,ADC类药物似乎更有可能获得成功。
一个靶点多个modality的探索在实体肿瘤中很常见,例如胃癌中CLDN18.2就是个案例。
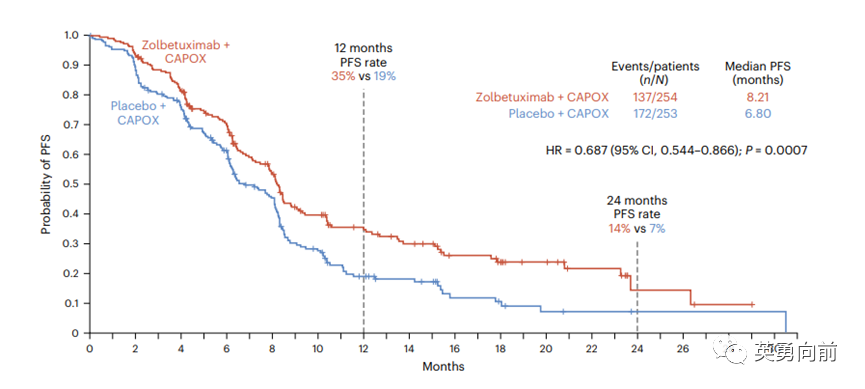
Zolbetuximab是靶向CLDN18.2的IgG1型单克隆抗体,GLOW全球三期研究(徐瑞华教授为全球PI)对比Zolbetuximab和安慰剂,联合卡培他滨和奥沙利铂(CAPOX)治疗初发胃癌/胃食管交界癌患者,1:1随机507例患者。

研究主要终点PFS分别为8.21个月和6.80个月(HR
0.687,P=0.0007);OS分别为14.39个月和12.16个月(HR
0.771,P=0.0118),安全性无差异。
GLOW研究连同2022年11月公布的SPOTLIGHT研究(联合mFOLFOX6),将支持Zolbetuximab的获批,PDUFA为2024年1月24日。
Zolbetuximab之后的各种modality探索也是热闹的很,从单抗到双抗、TCE、ADC、CAR-T百花齐放,超过20种。

在这篇发表于2023年9月NRDD的评论文章中,引用我国沈琳教授的一句话:“there are too many”。
有意思的是,作者也批判性地指出,对于胃癌等疾病,早期诊断治疗5年OS高于95%,而积极治疗HP的感染也将明显降低25%的胃癌发病率。
然而,我们仍然在不断堆积R&D的花费,这是创新的代价么?或许可以换个思路。

创新药往往从晚期开始逐步往早期推进,接下来介绍三篇发表在于NEJM关于NSCLC的研究结果。
7月13日,ADAURA研究最终数据公布,针对EGFR+的IB-IIIA期NSCLC患者,682例患者随机接受奥希替尼和安慰剂作为辅助治疗。
对于II到IIIA期,5年OS分别为85%和73%(HR 0.49),含I期总体人群的5y-OS为88%和78%(HR 0.49)。

8月10日,KEYNOTE-671研究发布结果。针对早期可切除患者,对比Pembro和安慰剂用于围手术期,研究入组397例患者。
中位随访25.2个月,两组的2y-EFS分别为62.4%和40.6%(HR 0.58),两组2y-OS分别为80.9%和77.6%(P=0.02)。

8月10日,NADIM II研究公布数据。这是一项临床II期研究,针对III期NSCLC患者新辅助Nivolumab联合化疗或单化疗,后手术,主要终点为病理学CR,次要终点为PFS何OS。
研究2:1入组86例患者,病理学CR分别为37%和7%,手术率为93%和69%,2y-PFS分别为67.2%和40.9%,2y-OS为85%和63.6%。

文章最后,介绍一篇关于ADC类药物开发的综述。文章由Aarvik Therapeutics公司撰写,于2023年8月28日发表于mABS杂志。

作者归纳至2022年11月,共260个ADC产品进入临床,其中11款产品获批,164个产品在研中,92个产品停止开发。
如果算下成功率,目前仅为4%,对比肿瘤药物开发成功率的平均水平不超过10%,我个人预估164个在研中,还能出来15个成功产品。当然,猜测没有依据,但确定性的是药物开发高失败率。
因此,何时决定停止开发以及判断依据,比继续开发更为重要。
本位归纳停止开发的三原因:
-不可耐受的毒性
-疗效不足
-商业考虑
大约1/3的产品由于毒性问题停止,包括:
1. 选择的靶点分布于正常细胞,或抗原表达过高导致必须使用高效payload导致的毒性。
2. 由于linker不稳定导致提前释放毒素。
3. ADC的胞饮作用。
4. Payload转化为毒性更大的代谢物。
例如,靶向CD44v6的药物Bivatuzumab,这款药物由靶向CD44抗人CD44v6抗体和DM1作为Payload组成,由于CD44v6同样表达于人体皮肤表面角质层,因此早期低剂量出现严重危及生命的皮肤脱落情况。
从Payload角度看,微管抑制剂(如auristatin衍生物)占所有失败案例的63%,其次是DNA合成抑制剂,而拓扑异构酶I抑制剂相对少。
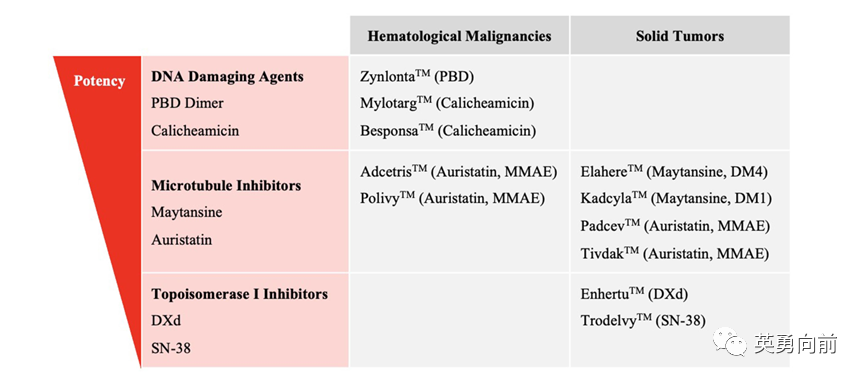

因此,开发一款成功的ADC需要天时地利人和。合适的靶向抗原,antibody-payload-linker的最佳组合,临床开发路径和运气。
版权声明:本网站所有注明来源“医微客”的文字、图片和音视频资料,版权均属于医微客所有,非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源:”医微客”。本网所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,转载仅作观点分享,版权归原作者所有。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 本站拥有对此声明的最终解释权。






































 关注公众号
关注公众号 安卓客户端
安卓客户端










发表评论
注册或登后即可发表评论
登录注册
全部评论(0)