
2023-09-13 来源 : JOJO的笔记本

一、竞争格局分析
最近有好好学习,越学越觉得自己什么都不会 ,差点就没有勇气写完这个合集了。不过咱本来就是个学习笔记,疏误难免,重在求知的乐趣。
,差点就没有勇气写完这个合集了。不过咱本来就是个学习笔记,疏误难免,重在求知的乐趣。
比如,我所说的“新药中的仿制药”,按照《注册管理办法》和大众认知,都属于中国式新药,不是《注册管理办法》和集采定义的“仿制药”,自己回看内容是有点绕。但是,凡事总有但是,绝大多数me better/me too新药,在我心里就是仿制药啊,这就是一不留神把心里实话说了出来 。
。
如果是真正的创新药,那么竞争格局分析过程不要太愉快,才不会动辄竞品信息多到一页PPT排版都艰难。连排版都艰难,新药开发会容易么?
竞争格局分析是个知己知彼的过程。
我们首先要非常清楚自家产品的最关键特征,才能对照着找出所有具备类似特征的竞品,尤其是自家产品对标&参照的竞品。还要非常清楚自家产品的临床开发计划和实际进展,才能和竞品们对弈。
当然,计划往往赶不上变化,未上市的新药,谁家不是赌。我们总是会根据竞争格局分析结果调整临床开发计划,也会根据临床开发实际进展重排竞争格局,这本来就是个漫长的博弈,一个季度更新一次版本也不稀奇。
竞品:可能对本产品构成竞争、瓜分市场份额的药物和新药产品。一个适应症甚至治疗领域的药物和新药产品是那么那么多,一定要挑出真正的竞品,而不是列举所有疗法。
个人建议的竞品纳入标准:
当然,如果已有竞品上市,或者关键性临床试验已发布结果,我们可以直接进行终极的疗效和安全性数据PK了。
竞品的最关键特征、临床开发计划和实际进展:又是评估参数如何获得的问题。绝大多数情况下,这些信息可以从竞品开发机构官网、学术会议报告、各国临床试验登记平台获得。尤其是前几年,但凡是个制药公司,总要跟投资者交代产品卖点和研发进展,抄作业总是那么轻松。这几年fast follow太凶猛,大家越来越舍不得剧透了,只想按法律规定的最低要求公开有限信息,所以现在做分析总要发挥人脉和经验猜一猜。
竞争格局分析的形式可以千变万化,我们只追求对所有评估参数一目了然,可以清晰比较出本产品 vs 竞品 vs 标准治疗SOC的竞争力(估算临床实践变更趋势和市场份额)。和产品生命周期管理目标一致,先按至少10年来估算。为了最后的PPT,我常用甘特图和房子图。
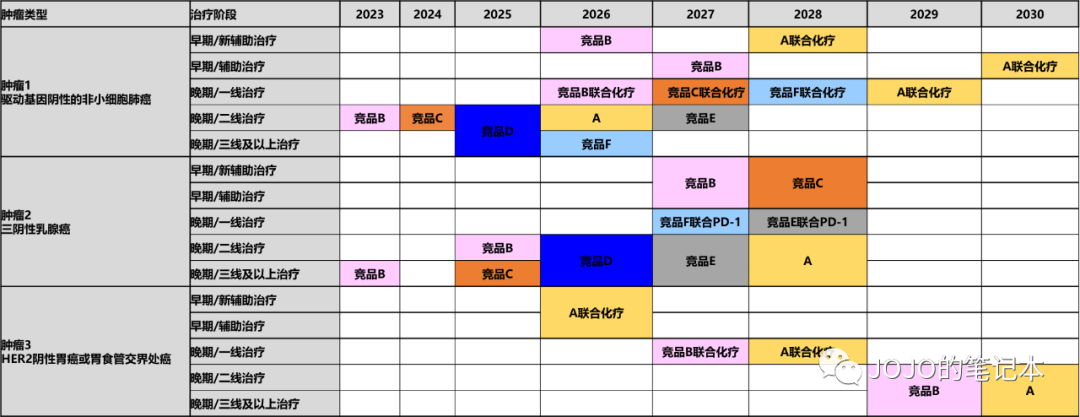
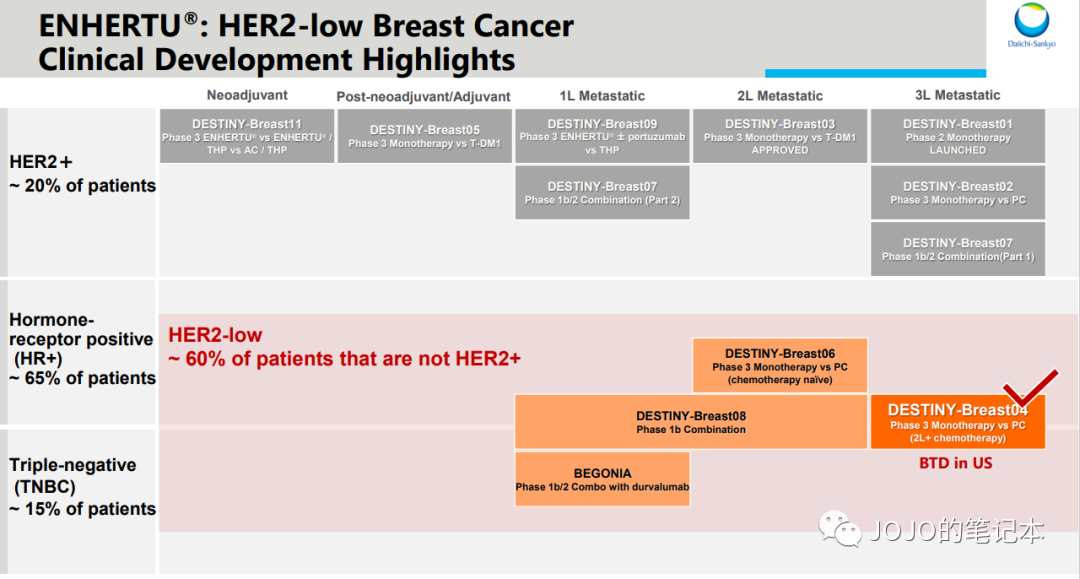
但是,凡事总有但是,我们还能产品线组合。或许Enhertu在部分适应症里不会直接取代HER2 TKI,而是联合HER2 TKI来取代HER2 TKI单药呢?HER2 TKI的产品生命周期又可以延续了。
二、临床策略开发
前面几个部分,都是拿着既定的临床开发策略给新药估值,如果估算结果是有价值但不多,还能优化么?如果我们正在以价值为导向制定临床开发策略,如何追求新药价值最大化?这就又回到了这张图。
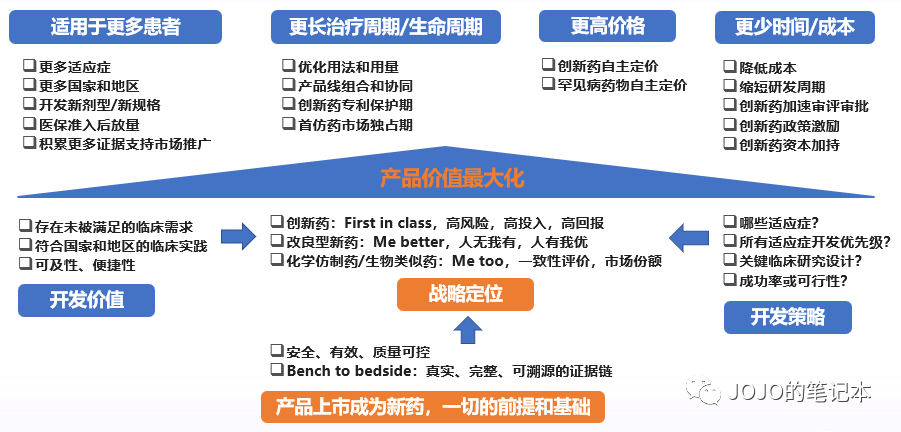
临床开发策略评估,绝不仅局限于临床试验成功率评估。
迟早,大家都会面对至少以下几个灵魂拷问:
1,至少要做哪些临床试验能证明产品安全、有效、质量/风险可控?
2,临床试验做完就一定会成功?(科学性、可行性、成本和周期)
3,临床试验成功就一定能上市?(监管机构的原则和基本要求)
4,新药上市就一定能有利润?(未被满足的临床需求、竞争格局)
这部分的重点是,产品获批上市成为新药,是一切价值的前提和基础。
所有既定临床试验都成功了,就一定能上市么?
举个栗子。2023年6月12日,嘉和生物发布公告,杰诺单抗用于治疗复发/难治性外周T细胞淋巴瘤(PTCL)的新药上市申请未获批准。这是国内首次PD-1上市申请被拒绝,这还是首个在国内甚至全球申请PTCL适应证的PD-1,2020年还被纳入优先审评名单。嘉和生物的解释是,PTCL有超过20种亚型,各亚型发病机制非常复杂,部分亚型发病机制尚不明确,目前全球没有PD-1产品被批准用于PTCL治疗,所以CDE审评极其谨慎。但业内更普遍认为,我们真的不需要更多PD-(L)1单抗了。
还有一些很特殊的情况,会导致临床试验成功但新药上市申请未获批准。比如“722惨案”之前的老产品,如果没被抛弃,很多都重做关键性临床试验了。医药行业基建之初,数据造假有但真是极少数,绝大多数撤回是没有按最高和最新的标准要求自己,很多关键性临床试验并不能够提供符合监管基本要求的真实、完整、可溯源的证据链。
还有一些很尴尬的情况,比如关键性临床试验进度太慢,导致成功时适应症的标准治疗方案已变更,或不再符合加速审批条件。中国式新药最容易遇上这种问题了。
继续举栗子,2021年之前胃癌三线治疗本没有靶向治疗甚至SOC。所以荣昌生物的RC48用于胃癌三线治疗,符合《单臂临床试验用于支持抗肿瘤药上市申请的适用性技术指导原则》的基本要求,可以凭借单臂临床试验获得有条件批准上市,上市后再做一个与医生选择的化疗头对头比较的随机、对照、验证性临床试验。那么其它HER2 ADC,如果选了同一个适应症也做单臂临床试验,即使不劣于RC48,试验完成时间晚于RC48获批时间怎么办?已经不再满足指导原则的基本要求了,即使成功,大概率只能再开展随机、对照临床试验用于申报。万一之后RC48验证性临床试验又成功了,获得完全批准成为新SOC,患者恐怕就不愿意入组化疗对照组了......或许,小概率CDE又在RC48之后多批了一个HER2 ADC用于胃癌三线治疗的单臂临床试验,但没有和化疗和RC48直接对比的数据,上市后市场推广的故事也不太好讲......
产品的战略定位。
我个人的理解,自家产品的卖点,或者说差异化优势,就是产品定位,也是临床开发策略的目标。以终为始,根据TPP制定临床开发策略,也是常用的产品管理方法。好卖的新药,总得有至少一方面显著优势,或者独家,或者首家,或者疗效更好,或者起效更快,或者毒副作用更轻更少,或者用药更便捷,都没有?那就只能说价格优势了。
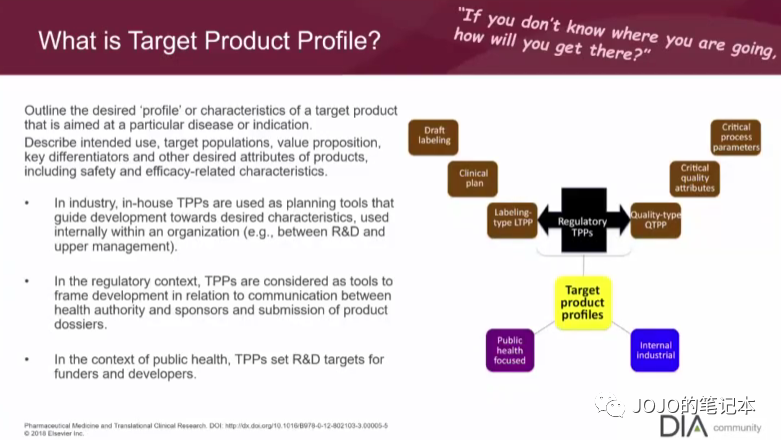
比如我们预判自家产品是首家,first in class,那我们的临床开发策略必须保证自家产品最快上市路线。比如我们预判自家产品疗效或安全性是同类最优,best in class,那必须有积累足够证据。
当然这些预判都是基于自家产品不断积累的数据和证据,TPP并不是一上来就非常清晰或一成不变的。总的有起点和终点,才能规划路线。
适用于更多患者,或更长的产品生命周期。
想要更多患者流和销售额,最直接就是开拓更多适应症。
基于自家产品的作用机制,列出所有可能的适应症,之后排序主次缓急。比如肿瘤靶向药,先看哪些肿瘤高表达这个靶点,再看针对这个靶点已经有多少种药物类型,最后看所有类型及本产品同类型已开发过哪些适应症。如果可选适应症太多,优先考虑本产品同类型已进入II期临床试验阶段的适应症,确定性高;也可以不走寻常路,专拣前人失败或无人区路线。
有些产品虽然适应症有限,但也可能在有限患者流里自我迭代,不断延长产品生命周期。比如先被批准用于成年人,后推广至儿童;先被批准静脉注射剂型,后开发皮下注射剂型;先被批准单药治疗,后联合其他药物治疗。
就算适应症有限,剂型和规格有限,我们还可以拓展至更多国家和地区。
思路还可以再开阔一点,也不是所有适用的疾病都必须尽快申报适应症。作用机制、疗效或安全性不那么确切?也可以先做一些IIT研究积累数据,之后或转为注册研究进一步开发,或直接市场推广。临床试验成本太高不堪重负?RWE也越来越被监管机构认可,也可以用于市场推广。
更短的临床开发周期。
国内外监管机构都有新药加速审评审批路径,一年半载的时间还是能节约出来的,有时候是不是首家/首仿,就差这一年半载。
现在再严格按I、II、III期划分临床试验也不太合适。我们必须通过临床试验来探索产品用于人体的药理、药代、剂量-效应-毒副反应关系,药物之间相互作用,用于具体适应症的疗效和安全性,这些探索可以在整个临床开发过程中持续完成,并不一定严格分期。这些年很多新颖的临床试验设计,seamless/lead-in/adaptive/umbrella/basket/platform/......都能显著缩短临床开发周期,监管机构也认可和提倡。
更低的临床开发成本。
行业内卷至此至今,真没有什么低垂果实可以轻易采摘了。如果一路立项评估下来没什么要紧问题,竞争格局分析时却几乎没有竞品,小心前面可能有大坑。我个人的经验,在国内这个大坑通常是不符合国内临床实际,或者开发成本太高甚至高过了预期总利润。
比如最早时候的细胞治疗和基因治疗,个体化治疗模式的成本高、单价高,国内市场很难推广。所以大家都在攻克规模化生产问题,以期降低成本,或者先走向欧美发达国家市场。
比如全新的靶向药,靶点筛查不属于国内临床常规项目,需要同步开发伴随诊断。
比如真正的创新药,所有质量标准等都没有现成的,都要独立开发。
比如肿瘤早期围手术期治疗,伦理通常会额外要求申办方负担患者手术甚至放疗费用。
比如主要终点指标涉及中心影像/病理,甚至中心ECG。
比如一些长病程疾病,现有标准治疗的疗效已经非常好,在此基础上进一步想获得有统计学意义的改善,需要几千例甚至上万例样本量。
更高的临床试验成功率。
总的来说,我建议按监管机构的最高和最新标准来要求自己。医药行业基建时代已经过了,新药就得有差异化优势,否则即使侥幸过了监管机构底限,还有市场竞争终极考验呢。
中国式新药,虽然主打一个对标原研药的验证,临床试验方案一般也不能照搬。时代不同,审评要求不同、临床实际不同,有时候还有人种差异。
患者入选/排除标准:范围太窄,患者数量太少;范围太宽,影响患者获益的因素太多,趋势无法保证。
统计假设参数:上限要现实,下限要符合监管机构最低要求。双终点和期中分析也不是万能的,α值不能过度消耗。
终点:抗肿瘤新药虽然可以采用pCR/ORR/PFS等替代终点作为主要终点,但至少CDE总想看看OS获益趋势(不一定是统计学差异)。有些疾病,有些新药类型,政策法规会额外要求长期安全性监测。
国内外同步开发:确认人种和疾病诊疗没有国别差异,能实施统一的诊断标准、终点评价标准、本产品用法用量、伴随治疗。否则还是分别开发。
I期临床试验:确认入组以后计划开发的所有适应症的患者,且用法用量最优。以前是真遇到过,传统3+3剂量爬坡推出的RP2D后期必须进一步优化;现在也是真遇到过,计划开发肿瘤ABC,I期临床试验入组时所有实体瘤都可以,入了一堆肿瘤DEF,没有数据支持后续开发。
产品线组合。
单用一个药物作为治疗方案的情况多么?虽然我们开发抗肿瘤新药,总是从末线患者单药治疗开始,但只要能耐受毒副作用,谁不想进一步联合其他药物增强疗效&克服耐药?二药、三药、四药.....同步、序贯、交替......
要考虑5-10年内所有治疗组合可能,提早布局。自家产品线丰富,联合自家其他产品最好;自家产品管线简单,积极拓展对外合作共赢。
因为,最后,医生和患者只会选择最优的那个治疗方案。
三、回到最初立项
这部分应该是新药估值方法学系列最后一PART了,后面有时间我再开其他合集闲扯淡,学海无涯。
有趣的是,在政策引导、行业公认、公司愿景、领导要求、自我洗脑等多重外在压力和内在动力的作用下,我自己突然想质疑一下。
只有开发创新药这一条生路么?仿制药就一点生存空间都没有了么?
我碰巧看到了一组数据,赫赛汀,最经典的HER2单抗,国内某家赫赛汀生物类似药的销售额,远高于国内另一家同款但不完全类似的HER2单抗创新药的销售额。
原因也不复杂,很好分析。生物类似药第一个适应症获批上市后,可以外推至原研药其他适应症。外推条件包括临床相关作用机制、PK、PD、有效性、安全性及免疫原性无显著差异,HER2靶点比较容易外推。
而HER2单抗创新药,最初为了避开专利,或进一步改善疗效和/或安全性,改造了分子结构,不符合生物类似药定义,只能归类到创新药。原研药那么经典,几千几万个分子里优选出来,后来的创新药稍微改造一下分子结构就能超越么?不劣于就很不错了,最后也未见得真在临床疗效和/或安全性上优于赫赛汀。但作为创新药,临床开发时只能一个个细分领域做关键性临床试验申报适应症。每提升一点患者流和销售额,对应的都是几倍临床开发成本。这一回合,中国式新药败给了生物类似药。
展开联想,那些最经典的适应症广泛的生物制品,美罗华(CD20单抗),贝伐珠(VEGF单抗),雷莫芦(VEGFR单抗)、西妥昔(EGFR单抗),阿达木单抗......是不是都会有类似情况?
我自己一直举的例子,Enhertu作为全新的HER2 ADC,显著优于之前所有HER2靶向治疗,就单药和单药疗效比较来说是的。但在某些适应症里,Enhertu单药不一定能优于THP方案(紫杉醇+赫赛汀+帕捷特),因为THP方案疗效和安全性都太出色,尤其是HP长期用药的安全性,Enhertu在此基础上几乎不可能做出进一步有统计学意义的提升,只能是提供另一种选择,不太可能完全取代。
继续质疑,新药开发一定要以终为始吗?
最近不是有个段子,“投资界宣称的技术突破太多、科技界已经跟不上了”。在投资界夜以继日、精益求精的勤奋工作下,我们构建了最科学的模型、完成了最全面的调查和最深入的分析,最终确定了最有市场价值的方向和亟待满足的临床需求,就差新药本身了。
终点的Flag都立好了,TPP也写好了,起点呢?新药呢?
很多疾病和靶点,因为技术层面的困难,不是不想开发,实在开发不出来。所以大家一起来天天吐槽中国式新药开发高度同质化导致恶性竞争,可是谁敢,或者说谁能开发真创新药?或许一开始大家并不是想内卷,就是很朴实地想有多大本事办多少事。
所以投资界都迷途知返了,这两年去投早研和技术平台,帮助解决技术瓶颈去了。
这里还可以多问一句,必须准备好目标产品特征(TPP)和临床开发计划(CDP)才能启动临床开发么?
TPP和CDP的概念来自外资big pharma,他们的实力和组织架构,需要且具备非常精细的决策机制,决策过程中控制风险最重要,计划不厌精,准备不厌细。内资公司和biotech?快速决策后灵活调整,野蛮生长好像也没问题。资源有限、时间更有限,方向差不多就可以出发了,一边赶路、一边观望、改进、补给。先上市再说。打个可能不太合适的比方,没条件正面战场大规模军团作战,游击战、闪电战.....都是战术。
如果回到最初的最初,实验室里立项呢?
拿着临床开发策略给新药估值,谁家不是赌。连临床开发策略都没有,在实验室里立项,赌得彻底,赌得纯粹。换个说法,这是一项非常非常具有前瞻性的工作。
我所向往的立项流程,大概就是前述新药估值流程,再往前面推一推,把临床前研发阶段和生产工艺验证阶段纳入评估。这些专业知识我早就还给了导师,不敢班门弄斧,只能稍微总结一下自己关注过问题。
(一)药物作用机制/疾病病理机制是否明确?
我个人还没有遇到过药物作用机制不明确的情况。但是总是有新的发现改变人们对疾病病理机制的认知,所以问题其实是,药物的作用机制,实际能对疾病病理机制造成多大影响。毕竟人体所有生理功能都是系统性的;正常生理和疾病状态是动态变化的;细胞内外信号传导是网不是线。
举个栗子。准新药临床前研究理论和实际都很顺利,POC临床试验里没获得预期疗效。从药物作用机制角度考虑可能的干扰包括:1)这个靶点如果是细胞内某个信号通路上的开关,是否这条信号通路的上下游还有别的开关?这边开那边关。2)这条信号通路之外,还有多少条分支/替代信号通路导向疾病进展?此路不通换条路。3)这个靶点有多少种配体?多个配体竞争上岗,准新药输。4)这个靶点如果是细胞膜表面蛋白,表达稳定么?会随着疾病进展增多么?会因为前序治疗起效减少么?肿瘤个体间和瘤内经常有异质性,进展过程中还能表型转换。5)组织内/细胞外微环境里,是否还存在其他调节机制?......
(二)目标靶点能成药吗?
以往新药开发的靶点总是激酶、受体或通道蛋白,需要获知精确的蛋白晶体结构,找到高亲和力和特异性的药物结合位点。能实现上述作用机制的靶点大概有几百种。据人类基因组学和蛋白质组学研究估计,目前仍有高达80%的疾病相关蛋白无法用药物调控。
比如,KRAS和SHP2曾经都被认为是“不可成药”的癌症靶点。
近几年一些新技术平台已经突破了上述限制。比如,蛋白靶向降解技术(PROTAC)、基因治疗和基因编辑等。
我觉得我没必要成为相关领域科学家,目前仍不可成药的靶点们,要么迟迟不能进入临床开发阶段、要么早期临床试验失败(迟迟不公开结果,也不是好信号)。还是那句话,行业内卷至此至今,真没有什么低垂果实可以轻易采摘了,无人区一定有无人的道理。
(三)产品可以规模化生产吗?
药品也是商品,需要在符合GMP要求的条件下大规模生产,才能品质稳定地广泛流通。很久以前的教科书上说,单抗药物生产工艺复杂,跟小分子化学药相比较,是造飞机和造自行车的区别。现在单抗药物生产工艺已经很成熟了,双/多抗、前药/纳米、ADC、PDC、XDY、核药、小核酸、PROTAC、T/NK/DC细胞......一个个都跟造火箭一样令人头秃。
即使规模化生产工艺建立并验证通过,原辅材料那边还有一堆卡脖子技术呢。包括但不局限于:高端检测仪器;生物反应器;特殊抗体、培养基、耗材、软件;核药的医用放射性同位素;siRNA的递送系统......
如果生产工艺中必须的原辅材料100%依赖进出口,谁知道什么时候就被卡了脖子,毕竟前几年为疫情所困,大家一起经历过lockdown。
(四)临床前药理毒理研究
产品分子/细胞结构上的差异化,有多少能转化为临床差异化?
临床前疾病模型:研究效率、复杂程度、临床相关性
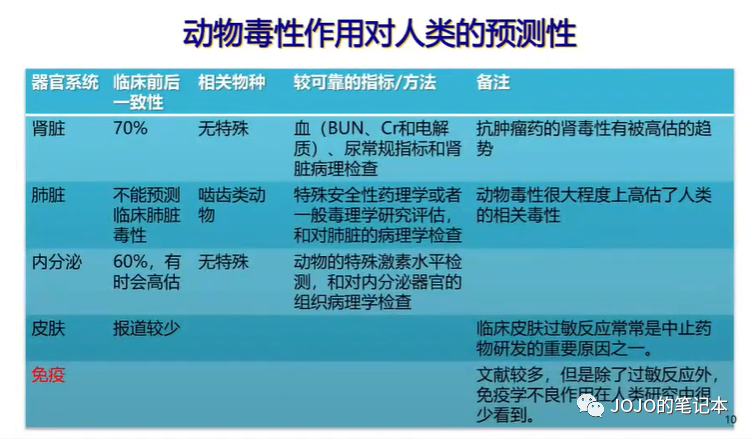
老友:这么多年了,猴子终于降价了。
我:建议知足,还有很多疾病至今没有药效模型。下图来自DIA。

(五)其他
知识产权纠纷:倒不至于阻碍新药上市,就是蛋糕要进一步切分。
MAH:听说而已,我没有亲见,听说我国跨境持有与生产的通道未打通?
(六)一些纯粹的吐槽,太长可不看
我常说,思路要再开阔一点。作为一个不太成熟的中年人,我总是希望,至少R&D类型的工作,不要有那么多刻板的偏见和限制。
我也算有一些经验,足够让我知道这世上唯一不变的就是变化,一切皆有可能。所以时刻提醒自己“什么都不懂+什么都不知情”,避免经验带来的偏见和标签,尽可能充分了解情况后,仅基于逻辑和证据做判断。
有个闻者伤心听者落泪的段子,我的本科专业是生命科学。至于怎么就在医药工业混了十五年,实验室、工厂、市场、运营、注册、医学部门统统都能混进去,主要是因为生活所迫、考研换专业到药学、赶上了医药行业基建时代。行业升级转型之快,我没有经验,别人也没有。新药开发本来就是最典型的系统工程,牵涉专业和领域之广,谁也不是全能的,大家不过是比拼学习方法和速度。
我的经验和过去的时代,都无法复制。就像在探险电影里,我所走过的路,前脚走后脚就坍塌,无迹可寻,无法回头。如果我真有经验可以分享,就是多看政策法规。国内CDE,国外FDA/EMA/TGA/PMDA/......还有ICH。反正我一直的感受,就是新药开发可能遇到的绝大多数问题,审评机构都给出过方向、路径、框架和原则。
不过现在大家都有了足够的经验和教训,都知道必须精通自家产品相关领域所有政策法规。起码在国内,CDE出品永远第一时间传诵,任何疑问都可以和CDE沟通交流,特殊情况下CDE也会与时俱进留有谈判空间,CDE可以和企业共建全新药物或技术的标准......当然,也永远都有“聪明”的人们灵活腾挪在法规限定范围内趋利避害,甚至在法规边缘疯狂试探。
版权声明:本网站所有注明来源“医微客”的文字、图片和音视频资料,版权均属于医微客所有,非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源:”医微客”。本网所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,转载仅作观点分享,版权归原作者所有。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 本站拥有对此声明的最终解释权。
































 关注公众号
关注公众号 安卓客户端
安卓客户端










发表评论
注册或登后即可发表评论
登录注册
全部评论(0)